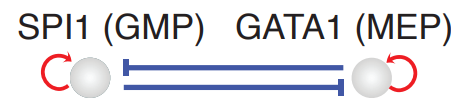
Molecular Interaction: Temporal Dominant Regulatory Interactions¶
To obtain mechanistic insights into key regulatory motifs from different perspectives, we developed three complementary strategies: cell-wise, trajectory-wise and plane-wise analyses. In this tutorial, we will introduce approaches for gene network motif analysis and guide you to perform cell-wise analyses of SPI1-GATA1 network motif.
Import relevant packages
import warnings
warnings.filterwarnings('ignore')
warnings.filterwarnings("ignore", message="numpy.dtype size changed")
import dynamo as dyn
dyn.configuration.set_figure_params('dynamo', background='white')
dyn.pl.style(font_path='Arial')
dyn.get_all_dependencies_version()
%load_ext autoreload
%autoreload 2
Using already downloaded Arial font from: /tmp/dynamo_arial.ttf
Registered custom font as: Arial
███ ████████
█████ █████ █████ █████ ███ █████
██████ ██████ ██████ ████████ ████
___ ████ ███
| \ _ _ _ _ __ _ _ __ ___ ███
| |) | || | ' \/ _` | ' \/ _ \█████ ███
|___/ \_, |_||_\__,_|_|_|_\___/█████ ████
|__/ ███ █████
Tutorial: https://dynamo-release.readthedocs.io/
█████
| package | umap-learn | typing-extensions | tqdm | statsmodels | setuptools | session-info | seaborn | scipy | requests | pynndescent | pre-commit | pandas | openpyxl | numdifftools | numba | networkx | mudata | matplotlib | loompy | leidenalg | igraph | dynamo-release | colorcet | anndata |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| version | 0.5.7 | 4.13.2 | 4.67.1 | 0.14.4 | 79.0.0 | 1.0.1 | 0.13.2 | 1.11.4 | 2.32.3 | 0.5.13 | 4.2.0 | 2.2.3 | 3.1.5 | 0.9.41 | 0.60.0 | 3.4.2 | 0.3.1 | 3.10.3 | 3.0.8 | 0.10.2 | 0.11.8 | 1.4.2rc1 | 3.1.0 | 0.11.4 |
adata_labeling = dyn.sample_data.hematopoiesis()
|-----> Downloading processed hematopoiesis adata
|-----> Downloading data to ./data/hematopoiesis.h5ad
|-----> File ./data/hematopoiesis.h5ad already exists.
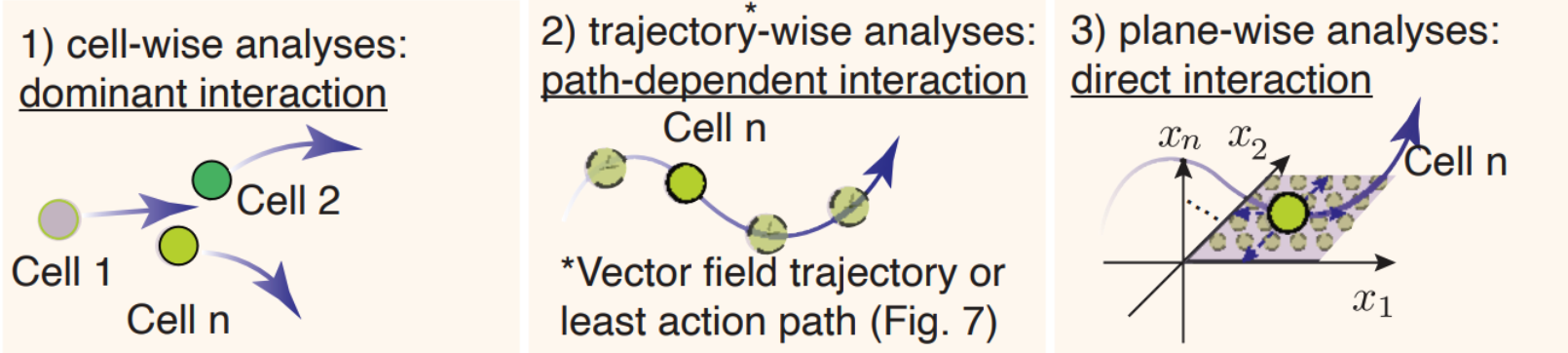
Three approaches for in-depth network motif characterizations¶
The schematic graph in this section shows the three approaches.
cell-wise analyses to reveal dominant interactions across all cells
trajectory-wise analyses reveal trajectory dependent interactions along a trajectory (predicted either from vector field streamline, or least action path, see Figure 6).
Plane-wise analyses reveal direct interactions for any characteristic cell states by varying genes of interest while holding all other genes constant.
In the next section, we will use cell-wise analyses to analyze PU.1/SPI1–GATA1 network motif.
Cell-wise analyses of the PU.1/SPI1–GATA1 network motif across all cells¶
We showcase cell-wise analyses with the canonical PU.1/SPI1-GATA1 network motif.
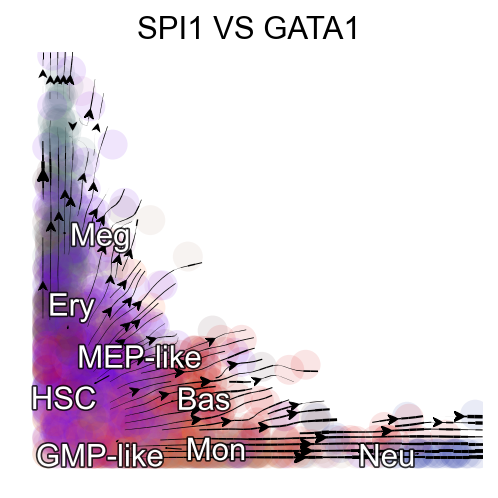
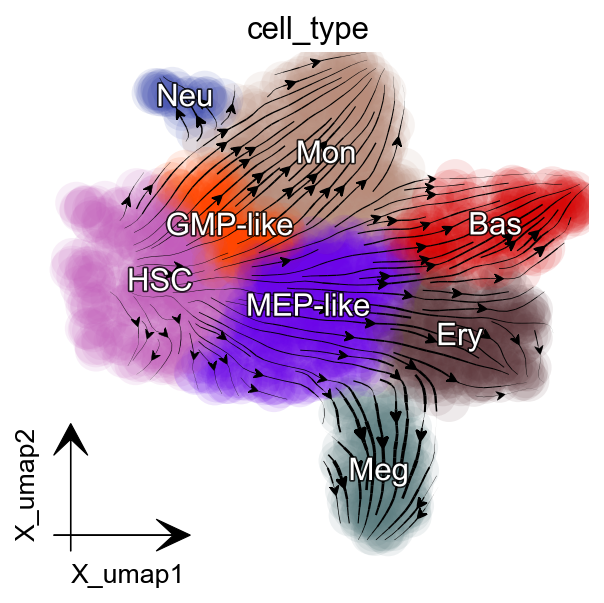
Streamline plot of the RNA velocities of SPI1 (x-axis) and GATA1 (y-axis)¶
The streamlines of SPI1 and GATA1 show that HSPCs bifurcate into GMP-like and MEP-like branches
#dyn.configuration.set_pub_style(scaler=4)
import matplotlib.pyplot as plt
fig, ax = plt.subplots(ncols=1, nrows=1,
constrained_layout=True,
figsize=(3, 3))
ax=dyn.pl.streamline_plot(
adata_labeling,
color="cell_type",
x="SPI1",
y="GATA1",
layer="M_t",
ekey="M_t",
pointsize=0.5,
ax=ax,
vkey="velocity_alpha_minus_gamma_s",
save_show_or_return='return',
s_kwargs_dict={'adjust_legend':True,'dpi':80,
'arrow':False},
)
|-----> method arg is None, choosing methods automatically...
|-----------> method kd_tree selected
|-----------> plotting with basis key=X_umap
|-----------> skip filtering cell_type by stack threshold when stacking color because it is not a numeric type
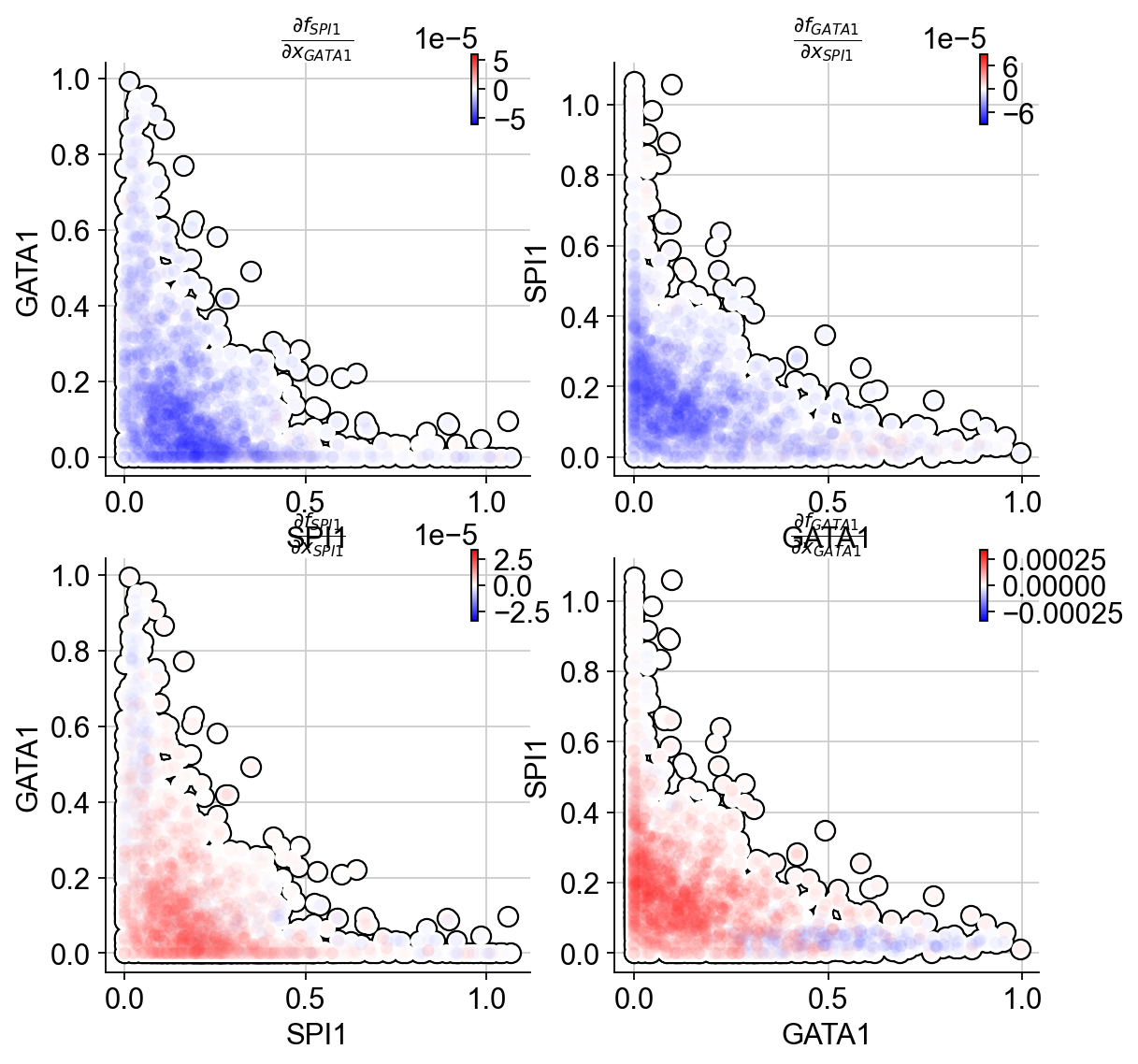
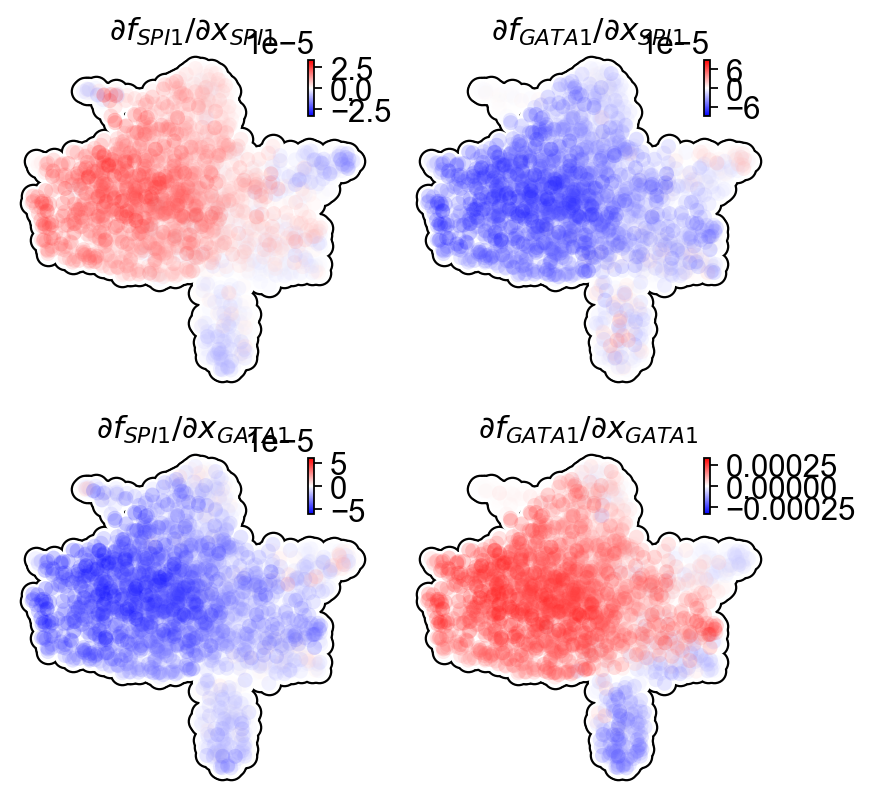
Next we will use jacobian to show
Repression from SPI1 to GATA1, GATA1 to SPI1
self-activation of SPI1, and GATA1, in the SPI1 and GATA1 expression space
In particular, the repression from SPI1 to GATA1 is mostly discernable in progenitors (rectangle A: bottom left) but becomes negligible when either GATA1 is much higher than SPI1 (rectangle B: upper left) or GATA1 is close to zero (rectangle C: bottom right).
%matplotlib inline
import numpy as np
genes = ["SPI1", "GATA1"]
def plot_jacobian_on_gene_axis(receptor, effector, x_gene=None, y_gene=None, axis_layer="M_t", temp_color_key="temp_jacobian_color", ax=None):
if x_gene is None:
x_gene = receptor
if y_gene is None:
y_gene = effector
x_axis = adata_labeling[:, x_gene].layers[axis_layer].A.flatten(),
y_axis = adata_labeling[:, y_gene].layers[axis_layer].A.flatten(),
dyn.vf.jacobian(adata_labeling, regulators = [receptor, effector], effectors=[receptor, effector])
J_df = dyn.vf.get_jacobian(
adata_labeling,
receptor,
effector,
)
color_values = np.full(adata_labeling.n_obs, fill_value=np.nan)
color_values[adata_labeling.obs["pass_basic_filter"]] = J_df.iloc[:, 0]
adata_labeling.obs[temp_color_key] = color_values
ax = dyn.pl.scatters(
adata_labeling,
vmin=0,
vmax=100,
color=temp_color_key,
cmap="bwr",
sym_c=True,
frontier=True,
sort="abs",
alpha=0.1,
pointsize=0.1,
x=x_axis,
y=y_axis,
save_show_or_return="return",
despline=True,
despline_sides=["right", "top"],
deaxis=False,
ax=ax,
arrow=False,
)
ax.set_title(r"$\frac{\partial f_{%s}}{\partial x_{%s}}$" % (effector, receptor))
ax.set_xlabel(x_gene)
ax.set_ylabel(y_gene)
adata_labeling.obs.pop(temp_color_key)
figure, axes = plt.subplots(2, 2, figsize=(8, 8))
plot_jacobian_on_gene_axis("GATA1", "SPI1", x_gene="SPI1", y_gene="GATA1", ax=axes[0,0])
plot_jacobian_on_gene_axis("SPI1", "GATA1", x_gene="GATA1", y_gene="SPI1", ax=axes[0,1])
plot_jacobian_on_gene_axis("SPI1", "SPI1", x_gene="SPI1", y_gene="GATA1", ax=axes[1,0])
plot_jacobian_on_gene_axis("GATA1", "GATA1", x_gene="GATA1", y_gene="SPI1",ax=axes[1,1])
|-----------> plotting with basis key=X_umap
|-----------> plotting with basis key=X_umap
|-----------> plotting with basis key=X_umap
|-----------> plotting with basis key=X_umap
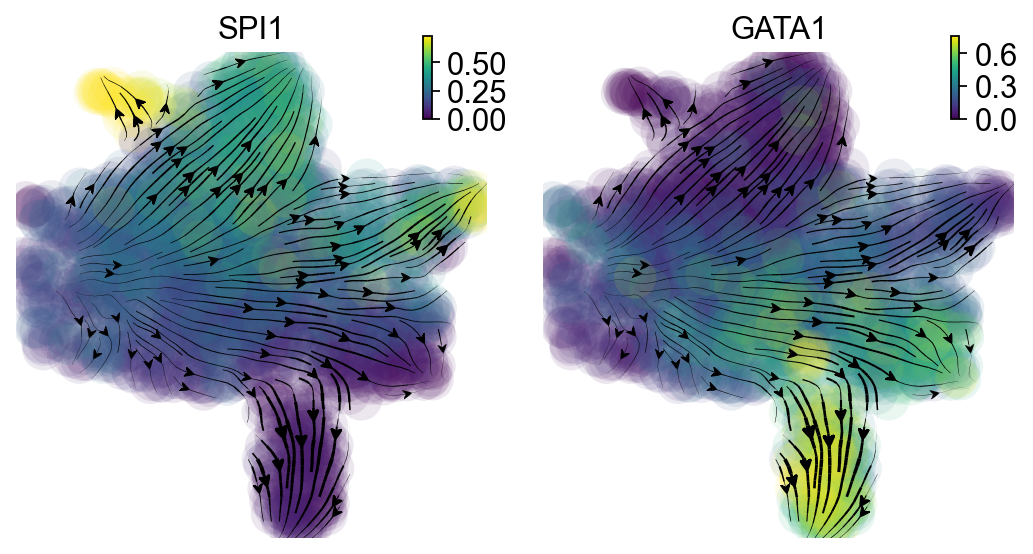
The streamlines of SPI1 and GATA1 in UMAP space and colored by M_t show that HSPCs bifurcate into GMP-like and MEP-like branches clearly.
dyn.pl.streamline_plot(
adata_labeling,
color=["cell_type"],
layer="M_t",
figsize=(4, 4),
ncols=2
)
dyn.pl.streamline_plot(
adata_labeling,
color=["SPI1", "GATA1"],
layer="M_t",
figsize=(4, 4),
ncols=2,
s_kwargs_dict={'adjust_legend':True,'dpi':80,
'arrow':False},
)
|-----> method arg is None, choosing methods automatically...
|-----------> method kd_tree selected
|-----------> plotting with basis key=X_umap
|-----------> skip filtering cell_type by stack threshold when stacking color because it is not a numeric type
|-----> method arg is None, choosing methods automatically...
|-----------> method kd_tree selected
|-----------> plotting with basis key=X_umap
|-----------> plotting with basis key=X_umap
UMAP jacobian analysis reveals self-activation of SPI1 in GMP and GATA1 in MEP, and mutual inhibition of SPI1 and GATA1 in GMP and MEP.
dyn.vf.jacobian(adata_labeling,
regulators = ["SPI1", "GATA1"])
dyn.pl.jacobian(adata_labeling, figsize=(3,3),
regulators = ["SPI1", "GATA1"])
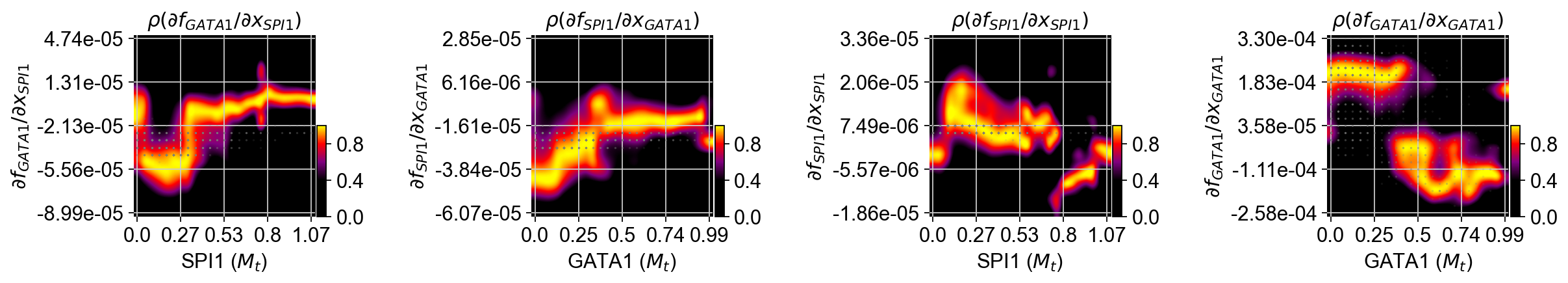
Response heatmap¶
White dashed lines indicate the minimum or maximum of repression or activation and the corresponding expression threshold.
%matplotlib inline
dyn.vf.jacobian(adata_labeling, regulators=["SPI1", "GATA1"],
effectors=["SPI1", "GATA1"])
dyn.pl.response(
adata_labeling,
np.array([["SPI1", "GATA1"], ["GATA1", "SPI1"],
["SPI1", "SPI1"], ["GATA1", "GATA1"]]),
ykey="jacobian",
log=False,
drop_zero_cells=True,
grid_num=25,
figsize=(4, 3),
save_show_or_return="show"
)
Conclusion¶
In the analyses above, we illustrate how to use dynamo to perform cell-wise analysis to explore the canonical PU.1/SPI1-GATA1 network motif. A schematic diagram of the SPI1-GATA1 toggle switch model can be summarized below.